Identification and verification of the ferroptosis- and pyroptosis-associated prognostic signature for low-grade glioma
DOI:
https://doi.org/10.17305/bjbms.2021.6888Keywords:
Ferroptosis, pyroptosis, prognostic signature, immunotherapy, low-grade gliomaAbstract
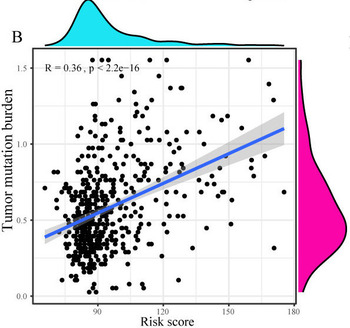
Accumulating evidence reveals that ferroptosis and pyroptosis play pivotal roles in tumorigenesis of low-grade glioma (LGG). In this research, we aimed to classify molecular subtypes and further identify and verify a novel multigene signature in LGG on the basis of ferroptosis and pyroptosis-related genes (FPRGs). Raw sequencing data and corresponding clinical data of LGG samples retrieved from the TCGA and CGGA databases were obtained for the training and validation datasets. Non-negative matrix factorization (NMF) clustering defined by FPRGs associated with prognosis was performed to classify molecular subtypes of LGG patients. LASSO-SVM-Random Forest analysis was carried out to develop an FPRG signature to predict the survival and benefit of immunotherapy of LGG patients. NMF clustering defined by FPRGs with prognostic values acted to categorize LGG patients into two molecular subtypes with different prognosis, clinical traits and immune microenvironments. A six-FPRG prognostic signature was constructed, accompanied by the optimal p-value. The AUC values of our signature exhibited great prognostic performances. Our signature was superior to other four well-recognized signatures in predicting the survival probability of LGG patients. Immune characteristics, tumor mutation profile, tumor stemness indices, MGMT methylation and immunotherapy response biomarkers showed significant differences between high- and low-risk populations. Finally, a nomogram was created for quantitative prediction of the survival probability of LGG patients, with the AUC values of the nomogram being 0.916, 0.888 and 0.836 for 1-, 3- and 5-year survival, sequentially. Overall, the FPRG signature may function as an effective indicator for the prognosis prediction and immunotherapy response of LGG patients.
Citations
Downloads
Downloads
Additional Files
Published
License
Copyright (c) 2022 Jie Wang, Jie Ren, Jifeng Liu, Linyun Zhang, Qihang Yuan, Bin Dong

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2022-03-05
Published 2022-09-16





