Attributes of intestinal microbiota composition and their correlation with clinical primary non-response to anti-TNF-α agents in inflammatory bowel disease patients
DOI:
https://doi.org/10.17305/bjbms.2021.6436Keywords:
Inflammatory bowel disease, non-responders, microbiotaAbstract
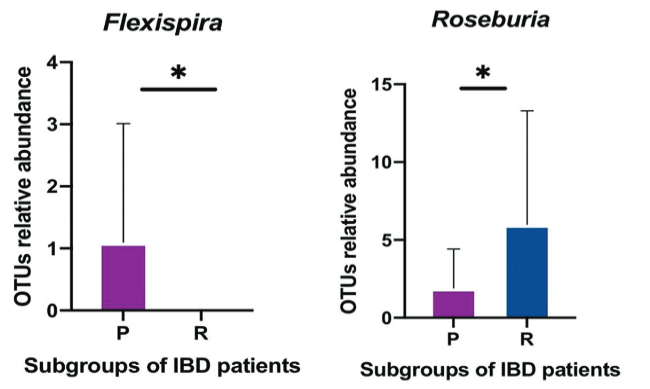
The largest microbial aggregation in the human body exists in the gastrointestinal tract. The microbiota in the host gastrointestinal tract comprises a diverse ecosystem, and the intestinal microbiota plays a vital role in maintaining gut homeostasis. This study aims to examine whether the gut microbiota influences unresponsiveness to anti-TNF-α treatments in primary nonresponder patients, and consequently identify the responsible microbes as biomarkers of unresponsiveness. Stool samples were collected from a cohort of patients with an established diagnosis of IBD, either ulcerative colitis (UC) or Crohn’s disease (CD), following completion of the induction phase of anti TNF therapy. 16S rRNA sequencing analysis was used to examine the pattern of microbiota communities in fecal samples. The quality and quantity of fecal microbiota were compared in responder and primary nonresponder IBD patients following anti-TNF-α therapy. As per our hypothesis, a difference in gut microbiome composition between the two patient subgroups was observed. A decreased abundance of short-chain fatty acid (SCFA)-producing bacteria, including Anaerostipes, Coprococcus, Lachnospira, Roseburia, and Ruminococcus, was detected in non-responsive patients, which was the hallmark of dysbiosis. Biomarkers of dysbiosis that were identified as predictors of clinical nonresponse, included Klebsiella, Eubacteriaceae, RF32, Bifidobacterium_animalis, and Muribaculaceae—previously known as S24-7. Signature biomarkers showed dramatic alteration in the composition of gut microbiota in patients who demonstrated primary nonresponse to anti-TNF-α agents. Dysbiosis, with features including a dropped biodiversity, augmentation in opportunistic pathogenic microbiota, and a lack of SCFA-producing bacteria, is a prominent feature of the microbiome of primary nonresponders to anti-TNF-α therapy.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2021 Hanan Alatawi, Mahmoud Mosli, Omar I. Saadah, Vito Annese, Rashad Al-Hindi, Marfat Alatawy, Hadba Al-Amrah, Dikhnah Alshehri, Ahmad Bahieldin, Sherif Edris

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2021-11-05
Published 2022-06-01





