Monomeric C-reactive protein affects cell injury and apoptosis through activation of p38 mitogen-activated protein kinase in human coronary artery endothelial cells
DOI:
https://doi.org/10.17305/bjbms.2020.4711Keywords:
C-reactive protein, cell injury, cell apoptosis, p38 mitogen-activated protein kinase, human coronary artery endothelial cellsAbstract
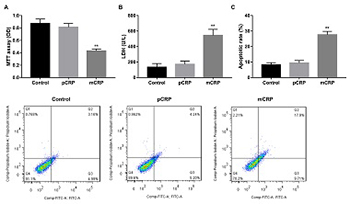
C-reactive protein (CRP) is an important predictor of cardiovascular events and plays a role in vascular inflammation and vessel damage. The aim of this study was to investigate the effect of pentameric CRP (pCRP) and monomeric CRP (mCRP) on the production of atherosclerosis-related factors in cultured human coronary artery endothelial cells (HCAECs). HCAECs were treated with pCRP, mCRP, p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580, or transfected with p38 MAPK siRNA. Western blotting was performed to detect the expression of vascular endothelial growth factor (VEGF), cyclooxygenase-2 (COX-2), intercellular adhesion molecule-2 (ICAM-2) and vascular cell adhesion molecule-1 (VCAM-1). Proliferation, damage, and apoptosis of HCAECs were examined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, lactate dehydrogenase (LDH), and flow cytometry, respectively. mCRP suppressed VEGF and COX-2 expression and enhanced ICAM-2 and VCAM-1 expression in HCAECs, in both dose-dependent and time-dependent manner. Except at 100 μg/ml concentration and 20-hour or 24-hour incubation, pCRP had no apparent effects. mCRP but not pCRP induced HCAEC injury and phosphorylation of p38 MAPK, and the inhibitor SB203580 reversed the effects of mCRP. mCRP promotes injury and apoptosis of HCAECs through a p38 MAPK-dependent mechanism, which provides a new therapy for the injury of HCAECs in atherosclerosis.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2020-03-31
Published 2020-11-02





