Craniosynostosis - Recognition, clinical characteristics, and treatment
DOI:
https://doi.org/10.17305/bjbms.2017.2083Keywords:
Craniosynostosis, development, classification, diagnosis, treatmentAbstract
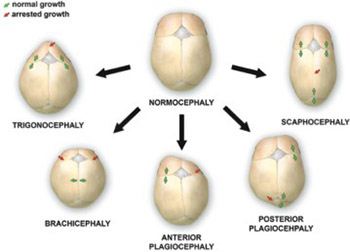
Craniosynostosis is a developmental craniofacial anomaly, resulting in impairment of brain development and abnormally shaped skull. The main cause of craniosynostosis is premature closure of one or more cranial sutures. It usually occurs as an isolated condition, but may also be associated with other malformations as part of complex syndromes. When left untreated, craniosynostosis can cause serious complications, such as developmental delay, facial abnormality, sensory, respiratory and neurological dysfunction, anomalies affecting the eye, and psychological disturbances. Thus, early diagnosis, expert surgical techniques, postoperative care, and adequate follow-up are of vital importance in treating craniosynostosis.
Citations
Downloads
References
Kjaer I. Neuro-osteology. Crit Rev Oral Biol Med 1998;9(2):224-44. https://doi.org/10.1177/10454411980090020501.
Chai Y, Maxson RE Jr. Recent advances in craniofacial morphogenesis. Dev Dyn 2006;235(9):2353-75. https://doi.org/10.1002/dvdy.20833.
Nah HD, Pacifici M, Gerstenfeld LC, Adams SL, Kirsch T. Transient chondrogenic phase in the intramembranous pathway during normal skeletal development. J Bone Miner Res 2000;15(3):522-33. https://doi.org/10.1359/jbmr.2000.15.3.522.
Gong SG. Cranial neural crest: Migratory cell behaviour and regulatory networks. Exp Cell Res 2014;325(2):90-5. https://doi.org/10.1016/j.yexcr.2014.03.015.
Kouskoura T, Fragou N, Alexiou M, John N, Sommer L, Graf D, et al. The genetic basis of craniofacial and dental abnormalities. Schweiz Monatsschr Zahnmed 2011;121(7-8):636-46.
Twigg SR, Wilkie AO. A Genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet 2015;97(3):359-77. https://doi.org/10.1016/j.ajhg.2015.07.006.
Zhao X, Qu Z, Tickner J, Xu J, Dai K, Zhang X. The role of SATB2 in skeletogenesis and human disease. Cytokine Growth Factor Rev 2014;25(1):35-44. https://doi.org/10.1016/j.cytogfr.2013.12.010.
Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: A brief review. Plast Reconstr Surg 2008;121(4):170e-8. https://doi.org/10.1097/01.prs.0000304441.99483.97.
van Adrichem LN, Hoogeboom AJ, Wolvius EB. Genetics of craniofacial development. [Article in Dutch]. Ned Tijdschr Tandheelkd 2008;115(2):61-8.
Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: Insights from human genetics and experimental studies. J Anat 2005;207(5):637-53. https://doi.org/10.1111/j.1469-7580.2005.00475.x.
Ogle RC, Tholpady SS, McGlynn KA, Ogle RA. Regulation of cranial suture morphogenesis. Cells Tissues Organs 2004;176(1-3):54-66. https://doi.org/10.1159/000075027.
Chim H, Manjila S, Cohen AR, Gosain AK. Molecular signaling in pathogenesis of craniosynostosis: The role of fibroblast growth factor and transforming growth factor-ß. Neurosurg Focus 2011;31(2):E7. https://doi.org/10.3171/2011.5.FOCUS1197.
Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn 2000;219(4):472-85. https://doi.org/10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F.
Delashaw JB, Persing JA, Jane JA. Cranial deformation in craniosynostosis. A new explanation. Neurosurg Clin N Am 1991;2(3):611-20.
Johnson D, Wilkie AO. Craniosynostosis. Eur J Human Genet 2011;19(4):369-76. https://doi.org/10.1038/ejhg.2010.235.
Sharma RK. Craniosynostosis. Indian J Plast Surg 2013;46(1):18-27. https://doi.org/10.4103/0970-0358.113702.
Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet A 2008;146A(8):984-91. https://doi.org/10.1002/ajmg.a.32208.
Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of nonsyndromic corronal craniosynostosis. Am J Med Genet 1995;55(4):500-4. https://doi.org/10.1002/ajmg.1320550422.
Cornelissen M, Ottelander BD, Rizopoulos D, van der Hulst R, Mink van der Molen A, van der Horst C, et al. Increase of prevalence of craniosynostosis. J Craniomaxillofac Surg 2016;44(9):1273-9. https://doi.org/10.1016/j.jcms.2016.07.007.
Sanchez-Lara PA, Carmichael SL, Graham JM Jr, Lammer EJ, Shaw GM, Ma C, et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet A 2010;152A(2):394-400. https://doi.org/10.1002/ajmg.a.33246.
Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 2010;126(2):e391-400. https://doi.org/10.1542/peds.2009-3491.
Carmichael SL, Ma C, Rasmussen SA, Honein MA, Lammer EJ, Shaw GM; National Birth Defects Prevention Study. Craniosynostosis and maternal smoking. Birth Defects Res A Clin Mol Teratol 2008;82(2):78-85. https://doi.org/10.1002/bdra.20426.
Gedzelman E, Meador KJ. Antiepileptic drugs in women with epilepsy during pregnancy. Ther Adv Drug Saf 2012;3(2):71-87. https://doi.org/10.1177/2042098611433192.
Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Semin Pediatr Neurol 2007;14(3):150-61. https://doi.org/10.1016/j.spen.2007.08.008.
Greenwood J, Flodman P, Osann K, Boyadjiev SA, Kimonis V. Familial incidence and associated symptoms in a population of individuals with nonsyndromic craniosynostosis. Genet Med 2014;16(4):302-10. https://doi.org/10.1038/gim.2013.134.
Kang SG, Kang JK. Current and future perspectives in craniosynostosis. J Korean Neurosurg Soc 2016;59(3):247-9. https://doi.org/10.3340/jkns.2016.59.3.247.
Saal HM. Genetic evaluation for craniofacial conditions. Facial Plast Surg Clin North Am 2016;24(4):405-25. https://doi.org/10.1016/j.fsc.2016.06.001.
Ryoo HG, Kim SK, Cheon JE, Lee JY, Wang KC, Phi JH. Slit ventricle syndrome and early-onset secondary craniosynostosis in an infant. Am J Case Rep 2014;15:246-53. https://doi.org/10.12659/AJCR.890590.
Cohen MM Jr. Sutural biology and the correlates of craniosynostosis. Am J Med Genet 1993;47(5):581-616. https://doi.org/10.1002/ajmg.1320470507.
Panchal J, Uttchin V. Management of craniosynostosis. Plast Reconstr Surg 2003;111(6):2032-48. https://doi.org/10.1097/01.PRS.0000056839.94034.47.
Esparza J, Hinojosa J, García-Recuero I, Romance A, Pascual B, Martínez de Aragón A, et al. Surgical treatment of isolated and syndromic craniosynostosis. Results and complications in 283 consecutive cases. Neurocirugia (Astur) 2008;19(6):509-29. https://doi.org/10.1016/S1130-1473(08)70201-X.
Governale LS. Craniosynostosis. Pediatr Neurol 2015;53(5):394-401. https://doi.org/10.1016/j.pediatrneurol.2015.07.006.
Zaleckas L, Neverauskienė A, Daugelavicius V, Šidlovskaitė-Baltakė D, Raugalas R, Vištartaitė B, et al. Diagnosis and treatment of craniosynostosis: Vilnius team experience. Acta Med Litu 2015;22(2):111-21. https://doi.org/10.6001/actamedica.v22i2.3126.
Tartaro A, Larici AR, Antonucci D, Merlino B, Colosimo C, Bonomo L. Optimization and diagnostic accuracy of computerized tomography with tridimensional spiral technique in the study of craniostenosis. [Article in Italian]. Radiol Med 1998;96(1-2):10-7.
Burokas L. Craniosynostosis: Caring for infants and their families. Crit Care Nurse 2013;33(4):39-50. https://doi.org/10.4037/ccn2013678.
Kim HJ, Roh HG, Lee IW. Craniosynostosis: Updates in radiologic diagnosis. J Korean Neurosurg Soc 2016;59(3):219-26. https://doi.org/10.3340/jkns.2016.59.3.219.
Stelnicki EJ, Mooney MP, Losken HW, Zoldos J, Burrows AM, Kapucu R, et al. Ultrasonic prenatal diagnosis of coronal suture synostosis. J Craniofac Surg 1997;8(4):252-8. https://doi.org/10.1097/00001665-199707000-00004.
Accardi MC, Lo Magno E, Ermito S, Dinatale A, Cacciatore A, Cavaliere A, et al. Echotomography of craniosynostosis: Review of literature. J Prenat Med 2009;3(2):31-3.
Miller C, Losken HW, Towbin R, Bowen A, Mooney MP, Towbin A, et al. Ultrasound diagnosis of craniosynostosis. Cleft Palate Craniofac J 2002;39(1):73-80. https://doi.org/10.1597/1545-1569(2002)039<0073:UDOC>2.0.CO;2.
Kosty J, Vogel TW. Insights into the development of molecular therapies for craniosynostosis. Neurosurg Focus 2015;38(5):E2. https://doi.org/10.3171/2015.2.FOCUS155.
Marie PJ, Kaabeche K, Guenou H. Roles of FGFR2 and twist in human craniosynostosis: Insights from genetic mutations in cranial osteoblasts. Front Oral Biol 2008;12:144-59. https://doi.org/10.1159/000115036.
Ciurea AV, Toader C. Genetics of craniosynostosis: Review of the literature. J Med Life 2009;2(1):5-17.
Katsianou MA, Adamopoulos C, Vastardis H, Basdra EK. Signaling mechanisms implicated in cranial sutures pathophysiology: Craniosynostosis. BBA Clin 2016;6:165-76. https://doi.org/10.1016/j.bbacli.2016.04.006.
Ursitti F, Fadda T, Papetti L, Pagnoni M, Nicita F, Iannetti G, et al. Evaluation and management of nonsyndromic craniosynostosis. Acta Peadiatr 2011;100(9):1185-94. https://doi.org/10.1111/j.1651-2227.2011.02299.x.
Shillito J Jr, Matson DD. Craniosynostosis: A review of 519 surgical patients. Pediatrics 1968;41(4):829-53.
Spazzapan P, Bosnjak R, Velnar T. Craniofacial reconstruction of the skull in anterior plagiocephaly: A case report. Br J Med Med Res 2016;18(2):1-7. https://doi.org/10.9734/BJMMR/2016/28075.
Haas-Lude K, Wolff M, Will B, Bender B, Krimmel M. Clinical and imaging findings in children with non-syndromic lambdoid synostosis. Eur J Pediatr 2014;173(4):435-40. https://doi.org/10.1007/s00431-013-2186-1.
Mulliken JB, Vander Woude DL, Hansen M, LaBrie RA, Scott RM. Analysis of posterior plagiocephaly: Deformational versus synostotic. Plast Reconstr Surg 1999;103(2):371-80. https://doi.org/10.1097/00006534-199902000-00003.
Aarnivala H, Vuollo V, Harila V, Heikkinen T, Pirttiniemi P, Holmström L, et al. The course of positional cranial deformation from 3 to 12 months of age and associated risk factors: A follow-up with 3D imaging. Eur J Pediatr 2016;175(12):1893-903. https://doi.org/10.1007/s00431-016-2773-z.
Speltz ML, Collett BR, Wallace ER, Starr JR, Cradock MM, Buono L, et al. Intellectual and academic functioning of school-age children with single-suture craniosynostosis. Pediatrics 2015;135(3):e615-23. https://doi.org/10.1542/peds.2014-1634.
Starr JR, Collett BR, Gaither R, Kapp-Simon KA, Cradock MM, Cunningham ML, et al. Multicenter study of neurodevelopment in 3-year-old children with and without single-suture craniosynostosis. Arch Pediatr Adolesc Med 2012;166(6):536-42. https://doi.org/10.1001/archpediatrics.2011.1800.
Chong S, Wang KC, Phi JH, Lee JY, Kim SK. Minimally invasive suturectomy and postoperative helmet therapy: Advantages and limitations. J Korean Neurosurg Soc 2016;59(3):227-32. https://doi.org/10.3340/jkns.2016.59.3.227.
Rottgers SA, Lohani S, Proctor MR. Outcomes of endoscopic suturectomy with postoperative helmet therapy in bilateral coronal craniosynostosis. J Neurosurg Pediatr 2016;18(3):281-6. https://doi.org/10.3171/2016.2.PEDS15693.
Jimenez DF, Barone CM. Endoscopic technique for sagittal synostosis. Childs Nerv Syst 2012;28(9):1333-9. https://doi.org/10.1007/s00381-012-1768-y.
Utria AF, Mundinger GS, Bellamy JL, Zhou J, Ghasemzadeh A, Yang R, et al. The importance of timing in optimizing cranial vault remodelling in syndromic craniosynostosis. Plast Reconstr Surg 2015;135(4):1077-84. https://doi.org/10.1097/PRS.0000000000001058.
Garza RM, Khosla RK. Nonsyndromic craniosynostosis. Semin Plast Surg 2012;26(2):53-63. https://doi.org/10.1055/s-0032-1320063.
Honeycutt JH. Endoscopic-assisted craniosynostosis surgery. Semin Plast Surg 2014;28(3):144-9. https://doi.org/10.1055/s-0034-1384810.
Esparza J, Hinojosa J. Complications in the surgical treatment of craniosynostosis and craniofacial syndromes: Apropos of 306 transcranial procedures. Childs Nerv Syst 2008;24(12):1421-30. https://doi.org/10.1007/s00381-008-0691-8.
Czerwinski M, Hopper RA, Gruss J, Fearon JA. Major morbidity and mortality rates in craniofacial surgery: An analysis of 8101 major procedures. Plast Reconstr Surg 2010;126(1):181-6. https://doi.org/10.1097/prs.0b013e3181da87df.
Downloads
Additional Files
Published
How to Cite
Accepted 2017-04-17
Published 2018-05-20






