The effect of metformin treatment on endoplasmic reticulum (ER) stress induced by status epilepticus (SE) via the PERK-eIF2α-CHOP pathway
DOI:
https://doi.org/10.17305/bjbms.2017.2044Keywords:
Apoptosis, CHOP expression, metformin, status epilepticus, SE, rat model, C/EBP homologous protein, eukaryotic initiation factor 2α, eIF2α, protein kinase RNA-like endoplasmic reticulum kinase, PERKAbstract
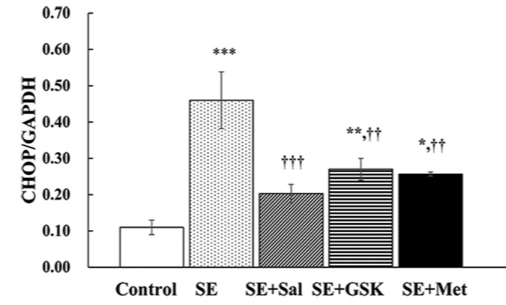
Status epilepticus (SE) is defined as continuous seizure activity lasting more than 5 minutes. It results in neuronal cell death, mediated by endoplasmic reticulum (ER) stress response. Previously, metformin demonstrated neuroprotective effects in primary cortical neurons. In this study, we analyzed the effect of metformin on ER stress via the pro-apoptotic protein kinase RNA-like endoplasmic reticulum kinase (PERK)-eukaryotic initiation factor 2α (eIF2α)-C/EBP homologous protein (CHOP) pathway. SE was induced in rats by pentylenetetrazole. Following SE, the rats were treated with salubrinal, GSK2656157, or metformin. In a control group (normal saline) SE was not induced. CHOP, eIF2α, and PERK expression was determined by Western blot; apoptosis was analyzed by TUNEL assay. CHOP expression was significantly increased at 6 and 24 hours following SE. At both time points, eIF2α and PERK levels were also increased. At 6 hours, CHOP expression was significantly reduced in salubrinal, GSK2656157 and metformin groups versus SE group. eIF2α and PERK levels were decreased in metformin compared to SE group. eIF2α expression was markedly decreased in salubrinal versus SE group, while PERK expression was markedly reduced in GSK2656157 versus SE group. At 6 and 24 hours, the apoptosis rate was significantly increased in SE versus control group, while it was significantly reduced in salubrinal, GSK2656157, and metformin groups compared to SE group. The apoptosis rate also decreased in salubrinal group at 24 hours, although not to the extent observed in metformin group. Overall, CHOP expression and apoptosis induced by SE in rats were reduced with metformin. Further studies are required to evaluate the clinical relevance of metformin for patients with SE.
Citations
Downloads
References
Sánchez S, Rincon F. Status epilepticus: Epidemiology and public health needs. J Clin Med 2016;5(8). pii:E71. https://doi.org/10.3390/jcm5080071.
DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46(4):1029-35. https://doi.org/10.1212/WNL.46.4.1029.
Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Incidence of status epilepticus in Rochester, Minnesota, 1965-1984. Neurology 1998;50(3):735-41. https://doi.org/10.1212/WNL.50.3.735.
Hui AC, Joynt GM, Li H, Wong KS. Status epilepticus in Hong Kong Chinese: Aetiology, outcome and predictors of death and morbidity. Seizure 2003;12(7):478-82. https://doi.org/10.1016/S1059-1311(03)00024-4.
Li JM, Chen L, Zhou B, Zhu Y, Zhou D. Convulsive status epilepticus in adults and adolescents of Southwest China: Mortality, etiology, and predictors of death. Epilepsy Behav 2009;14(1):146-9. https://doi.org/10.1016/j.yebeh.2008.09.005.
Nishiyama I, Ohtsuka Y, Tsuda T, Kobayashi K, Inoue H, Narahara K, et al. An epidemiological study of children with status epilepticus in Okayama, Japan: Incidence, etiologies, and outcomes. Epilepsy Res 2011;96(1-2):89-95. https://doi.org/10.1016/j.eplepsyres.2011.05.004.
Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol 2008;8(1):75-81. https://doi.org/10.1016/j.coph.2007.07.005.
Lopes MW, Soares FM, de Mello N, Nunes JC, Cajado AG, de Brito D, et al. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res 2013;226(2):153-63. https://doi.org/10.1007/s00221-013-3421-8.
Laurén HB, Ruohonen S, Kukko-Lukjanov TK, Virta JE, Grönman M, Lopez-Picon FR, et al. Status epilepticus alters neurogenesis and decreases the number of GABAergic neurons in the septal dentate gyrus of 9-day-old rats at the early phase of epileptogenesis. Brain Res 2013;1516:33-44. https://doi.org/10.1016/j.brainres.2013.04.028.
Pelletier MR, Wadia JS, Mills LR, Carlen PL. Seizure-induced cell death produced by repeated tetanic stimulation in vitro: Possible role of endoplasmic reticulum calcium stores. J Neurophysiol 1999;81(6):3054-64.
Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, et al. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuropathol Exp Neurol 2006;65(3):217-25. https://doi.org/10.1097/01.jnen.0000202886.22082.2a.
Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, et al. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 2007;27(4):901-8. https://doi.org/10.1523/JNEUROSCI.4289-06.2007.
Xu B, Shan M, Wang F, Deng Y, Liu W, Feng S, et al. Endoplasmic reticulum stress signaling involvement in manganese-induced nerve cell damage in organotypic brain slice cultures. Toxicol Lett 2013;222(3):239-46. https://doi.org/10.1016/j.toxlet.2013.08.001.
Chen J, Guo H, Zheng G, Shi ZN. Region-specific vulnerability to endoplasmic reticulum stress-induced neuronal death in rat brain after status epilepticus. J Biosci 2013;38(5):877-86. https://doi.org/10.1007/s12038-013-9391-y.
Berthier A, Payá M, García-Cabrero AM, Ballester MI, Heredia M, Serratosa JM, et al. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of Lafora disease. Mol Neurobiol 2016;53(2):1296-309.
https://doi.org/10.1007/s12035-015-9091-8.
Chung MM, Chen YL, Pei D, Cheng YC, Sun B, Nicol CJ, et al. The neuroprotective role of metformin in advanced glycation end product treated human neural stem cells is AMPK-dependent. Biochim Biophys Acta 2015;1852(5):720-31. https://doi.org/10.1016/j.bbadis.2015.01.006.
El-Mir MY, Detaille D, R-Villanueva G, Delgado-Esteban M, Guigas B, Attia S, et al. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci 2008;34(1):77-87. https://doi.org/10.1007/s12031-007-9002-1.
Jung TW, Lee MW, Lee YJ, Kim SM. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem Biophys Res Commun 2012;417(1):147-52. https://doi.org/10.1016/j.bbrc.2011.11.073.
Simon-Szabó L, Kokas M, Mandl J, Kéri G, Csala M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS One 2014;9(6):e97868. DOI: 10.1371/journal.pone.0097868.
Chen J, Zheng G, Guo H, Shi ZN. Role of endoplasmic reticulum stress via the PERK signaling pathway in brain injury from status epilepticus. J Mol Neurosci 2014;53(4):677-83. https://doi.org/10.1007/s12031-014-0236-4.
Lado FA, Sperber EF, Moshé SL. Anticonvulsant efficacy of gabapentin on kindling in the immature brain. Epilepsia 2001;42(4):458-63. https://doi.org/10.1046/j.1528-1157.2001.30900.x.
Algire C, Moiseeva O, Deschênes-Simard X, Amrein L, Petruccelli L, Birman E, et al. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res (Phila) 2012;5(4):536-43. https://doi.org/10.1158/1940-6207.CAPR-11-0536.
B'chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2a/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013;41(16):7683-99. https://doi.org/10.1093/nar/gkt563.
Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999;397(6716):271-4. https://doi.org/10.1038/16729.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000;5(5):897-904. https://doi.org/10.1016/S1097-2765(00)80330-5.
Chen YJ, Su JH, Tsao CY, Hung CT, Chao HH, Lin JJ, et al. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and PERK/eIF2α/ATF4/CHOP pathway. Molecules 2013;18(9):10146-61. https://doi.org/10.3390/molecules180910146.
Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci 2010;30(50):16938-48. https://doi.org/10.1523/JNEUROSCI.1598-10.2010.
Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2a-ATF4-CHOP signaling contributes to TNFa-induced vascular calcification. J Am Heart Assoc 2013;2(5):e000238. https://doi.org/10.1161/JAHA.113.000238.
Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death-mechanisms of ER stress-induced cell death. Biol Chem 2014;395(1):1-13. https://doi.org/10.1515/hsz-2013-0174.
Lopez-Meraz ML, Niquet J, Wasterlain CG. Distinct caspase pathways mediate necrosis and apoptosis in subpopulations of hippocampal neurons after status epilepticus. Epilepsia 2010;51(Suppl 3):56-60. https://doi.org/10.1111/j.1528-1167.2010.02611.x.
Fatt M, Hsu K, He L, Wondisford F, Miller FD, Kaplan DR, et al. Metformin acts on two different molecular pathways to enhance adult neural precursor proliferation/self-renewal and differentiation. Stem Cell Reports 2015;5(6):988-95. https://doi.org/10.1016/j.stemcr.2015.10.014.
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2017 Bosnian Journal of Basic Medical Sciences

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2017-05-27
Published 2018-02-20





