TAFRO syndrome: New subtype of idiopathic multicentric Castleman disease
DOI:
https://doi.org/10.17305/bjbms.2017.1930Keywords:
TAFRO, Castleman disease, lymphoproliferative disorders, interleukin-6, cytokines, cytokine stormAbstract
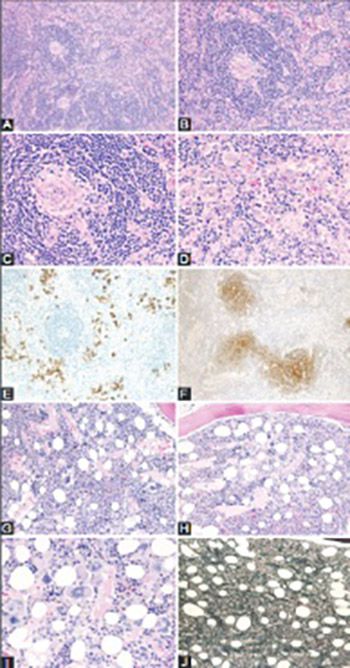
Castleman disease (CD) describes a group of three rare and poorly understood lymphoproliferative disorders that have heterogeneous clinical symptoms and common lymph node histopathological features. Unicentric CD (UCD) involves a single region of enlarged nodes. Multicentric CD (MCD) involves multiple regions of enlarged lymph nodes, constitutional symptoms, and organ dysfunction due to a cytokine storm often including interleukin 6. MCD is further divided into Human Herpes Virus-8 (HHV-8)-associated MCD, which occurs in immunocompromised individuals, and HHV-8-negative/idiopathic MCD (iMCD). Recently, iMCD has been further sub-divided into patients with TAFRO syndrome, which involves thrombocytopenia (T), anasarca (A), fevers (F), reticulin myelofibrosis (R), organomegaly (O), and normal or only slightly elevated immunoglobulin levels, and those who do not have TAFRO syndrome. Non-TAFRO iMCD patients typically have thrombocytosis, less severe fluid accumulation, and hypergammaglobulinemia. iMCD patients with TAFRO syndrome may have a worse prognosis, but more research is needed.
Citations
Downloads
References
Castleman B, Towne VW. Case records of the Massachusetts General Hospital: Case No. 40231. N Engl J Med 1954;250(23):1001-5. https://doi.org/10.1056/NEJM195406102502308.
Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer 1956;9(4):822-30. https://doi.org/10.1002/1097-0142(195607/08)9:4<822::AID-CNCR2820090430>3.0.CO;2-4.
Gaba AR, Stein RS, Sweet DL, Varikojis D. Multicentric giant lymph node hyperplasia. Am J Clin Pathol 1978;69(1):86-90. https://doi.org/10.1093/ajcp/69.1.86.
Bowne WB, Lewis JJ, Filippa DA, Niesvizky R, Brooks AD, Burt ME, et al. The management of unicentric and multicentric Castleman's disease: A report of 16 cases and a review of the literature. Cancer 1999;85(3):706-17. https://doi.org/10.1002/(SICI)1097-0142(19990201)85:3<706::AID-CNCR21>3.0.CO;2-7.
Waterston A, Bower M. Fifty years of multicentric Castleman's disease. Acta Oncol 2004;43(8):698-704. https://doi.org/10.1080/02841860410002752.
Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 1995;86(4):1276-80.
Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 1994;266(5192):1865-9. https://doi.org/10.1126/science.7997879.
Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996;274(5293):1739-44. https://doi.org/10.1126/science.274.5293.1739.
Suda T, Katano H, Delsoi G, Kakiuchi C, Nakamura T, Shiota M, et al.
HHV-8 infection status of AIDS-related and AIDS-associated multicentric Castleman’s disease. Pathol Int 2001;51(9):671-9. https://doi.org/10.1046/j.1440-1827.2001.01266.x.
Dossier A, Meignin V, Fieshi C, Boutboul D, Oksenhendler E, Galicier L. Human herpesvirus 8-related Castleman disease in the absence of HIV infection. Clin Infect Dis 2013;56(6):833-42. https://doi.org/10.1093/cid/cis1009.
El-Osta HE, Kurzrock R. Castleman's disease: From basic mechanisms to molecular therapeutics. Oncologist 2011;16(4):497-511.
https://doi.org/10.1634/theoncologist.2010-0212.
Faigenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood 2014; 123(19):2924-33. https://doi.org/10.1182/blood-2013-12-545087.
Fajgenbaum DC, Ruth JR, Kelleher D, Rubenstein AH. The collaborative network approach: A new framework to accelerate Castleman's disease and other rare disease research. Lancet Haematol 2016;3(4):e150-2. https://doi.org/10.1016/S2352-3026(16)00007-7.
Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma 2015;56(5):1252-60. https://doi.org/10.3109/10428194.2014.953145.
Talat N, Schulte KM. Castleman’s disease: Systemic analysis of 416 patients from the literature. Oncologist 2011;16(9):1316-24. https://doi.org/10.1634/theonclogist.2011-0075.
Peterson BA, Frizzera G. Multicentric Castleman’s disease. Semin Oncol 1993;20(6):636-47.
Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman's disease: A systematic literature review. Lancet Haematol 2016;3(4):e163-75. https://doi.org/10.1016/S2352-3026(16)00006-5.
Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017 (in press). https://doi.org/10.1182/blood-2016-10-746933.
Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. The clinical spectrum of Castleman's disease. Am J Hematol 2012;87(11):997-1002.
https://doi.org/10.1002/ajh.23291.
Waterston A, Bower M. Fifty years of multicentric Castleman's disease. Acta Oncol 2004;43(8):698-704. https://doi.org/10.1080/02841860410002752.
Fajgenbaum D, Rosenbach M, van Rhee F, Nasir A, Reutter J. Eruptive cherry hemangiomatosis associated with multicentric Castleman disease: A case report and diagnostic clue. JAMA Dermatol 2013;149(2):204-8.
https://doi.org/10.1001/jamadermatol.2013.1552.
Nishmoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005;106(8):2627-32. https://doi.org/10.1182/blood-2004-12-4602.
Ahmed B, Tschen JA, Cohen PR, Zaki MH, Rady PL, Tyring SK, et al. Cutaneous castleman's disease responds to anti interleukin-6 treatment. Mo Cancer Ther 2007;6(9):2386-90. https://doi.org/10.1158/1535-7163.mct-07-0256.
Zhai S, Simpson D. Polynesian variant of idiopathic multicentric Castleman disease [abstract]. Blood 2013;122(21):Abstract 5127.
Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. [Article in Japanese]. Rinsho Ketsueki 2010;51(5):320-5.
http://doi.org/10.11406/rinketsu.51.320.
Abdo LA, Morin CP, Collarino RP, Cabane JP, Gatfosse MA. First European case of TAFRO syndrome associated with Sjogren disease. Am J Intern Med 2014;2(6):102-5.
https://doi.org/10.11648/j.ajim.20140206.12.
Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J of Hematol 2016;91(2):220-6.
https://doi.org/10.1002/ajh.24242.
Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome): A novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: A status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop 2013;53(1):57-61. https://doi.org/10.3960/jslrt.53.57.
Masaki Y, Kawabata H, Takai K, Kojima M, Tsukamoto N, Ishigaki Y, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome. Int J Hematol 2016;103(6):686-92. https://doi.org/10.1007/s12185-016-1979-1.
Downloads
Additional Files
Published
How to Cite
Accepted 2017-01-14
Published 2017-05-20





