Time course of global gene expression alterations in Candida albicans during infection of HeLa cells
DOI:
https://doi.org/10.17305/bjbms.2017.1667Keywords:
Candida albicans, infection, differential gene expression, HeLa cellsAbstract
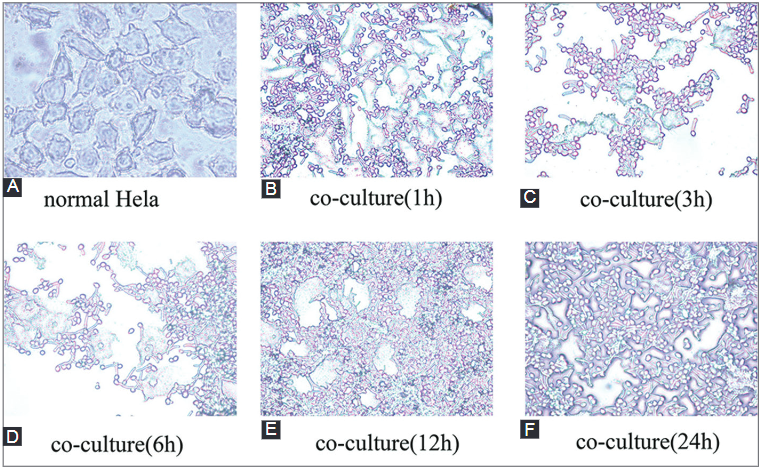
Candida albicans (C. albicans) is an opportunistic fungus that quickly adapts to various microniches. It causes candidiasis, a common fungal infection for which the pathogenic mechanism has not been elucidated yet. To explore the pathogenic mechanism of candidiasis we used several methods, including microscopic observation of morphological changes of HeLa cells and fungus, analysis of differentially expressed genes using gene chips, and a series of biological and bioinformatic analyses to explore genes that are possibly involved in the pathogenesis of C. albicans. During the C. albicans infection, significant morphological changes of the fungus were observed, and the HeLa cells were gradually destroyed. The gene chip experiments showed upregulated expression of 120 genes and downregulated expression of 178 genes. Further analysis showed that some genes may play an important role in the pathogenesis of C. albicans. Overall, morphological variation and adaptive gene expression within a particular microniche may exert important effects during C. albicans infections.
Citations
Downloads
References
Sobel JD. Vulvovaginal candidosis. Lancet 2007;369(9577):1961-71. https://doi.org/10.1016/S0140-6736(07)60917-9.
Sobel JD. Management of patients with recurrent vulvovaginal candidiasis. Drugs 2003;63(11):1059-66. https://doi.org/10.2165/00003495-200363110-00002.
Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol 2004;12(7):317-24. https://doi.org/10.1016/j.tim.2004.05.008.
Kabir MA, Hussain MA. Human fungal pathogen Candida albicans in the postgenomic era: An overview. Expert Rev Anti Infect Ther 2009;7(1):121-34. https://doi.org/10.1586/14787210.7.1.121.
Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, et al. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A 2004;101(19):7329-34. https://doi.org/10.1073/pnas.0401648101.
Cheng G, Wozniak K, Wallig MA, Fidel PJ, Trupin SR, Hoyer LL. Comparison between Candida albicans agglutinin-like sequence gene expression patterns in human clinical specimens and models of vaginal candidiasis. Infect Immun 2005;73(3):1656-63. https://doi.org/10.1128/IAI.73.3.1656-1663.2005.
Fradin C, De Groot P, MacCallum D, Schaller M, Klis F, Odds FC, et al. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol Microbiol 2005;56(2):397-415.
https://doi.org/10.1111/j.1365-2958.2005.04557.x.
Clarke R, Ressom HW, Wang A, Xuan J, Liu MC, Gehan EA, et al. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat Rev Cancer 2008;8(1):37-49. https://doi.org/10.1038/nrc2294.
Ramoni MF, Sebastiani P, Kohane IS. Cluster analysis of gene expression dynamics. Proc Natl Acad Sci U S A 2002;99(14):9121-6. https://doi.org/10.1073/pnas.132656399.
Lamanna AC, Karbstein K. An RNA conformational switch regulates pre-18S rRNA cleavage. J Mol Biol 2011;405(1):3-17. https://doi.org/10.1016/j.jmb.2010.09.064.
Yi M, Horton JD, Cohen JC, Hobbs HH, Stephens RM. WholePathwayScope: A comprehensive pathway-based analysis tool for high-throughput data. Bmc Bioinformatics 2006;7:30. https://doi.org/10.1186/1471-2105-7-30.
Nikiforova VJ, Willmitzer L. Network visualization and network analysis. Experientia Suppl 2007;97:245-75. https://doi.org/10.1007/978-3-7643-7439-6_11.
Kumamoto CA, Vinces MD. Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cell Microbiol 2005;7(11):1546-54.
https://doi.org/10.1111/j.1462-5822.2005.00616.x.
Calderone RA, Fonzi WA. Virulence factors of Candida albicans. Trends Microbiol 2001;9(7):327-35. https://doi.org/10.1016/S0966-842X(01)02094-7.
Naglik J, Albrecht A, Bader O, Hube B. Candida albicans proteinases and host/pathogen interactions. Cell Microbiol 2004;6(10):915-26.
https://doi.org/10.1111/j.1462-5822.2004.00439.x.
Wilson D, Thewes S, Zakikhany K, Fradin C, Albrecht A, Almeida R, et al. Identifying infection-associated genes of Candida albicans in the postgenomic era. Fems Yeast Res 2009;9(5):688-700.
https://doi.org/10.1111/j.1567-1364.2009.00524.x.
Zakikhany K, Naglik JR, Schmidt-Westhausen A, Holland G, Schaller M, Hube B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol 2007;9(12):2938-54.
https://doi.org/10.1111/j.1462-5822.2007.01009.x.
Ramage G, VandeWalle K, Lopez-Ribot JL, Wickes BL. The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. Fems Microbiol Lett 2002;214(1):95-100.
https://doi.org/10.1111/j.1574-6968.2002.tb11330.x.
Whiteway M, Oberholzer U. Candida morphogenesis and host-pathogen interactions. Curr Opin Microbiol 2004;7(4):350-7. https://doi.org/10.1016/j.mib.2004.06.005.
de Boer AD, de Groot PW, Weindl G, Schaller M, Riedel D, Diez-Orejas R, et al. The Candida albicans cell wall protein Rhd3/Pga29 is abundant in the yeast form and contributes to virulence. Yeast 2010;27(8):611-24. https://doi.org/10.1002/yea.1790.
Cappellaro C, Mrsa V, Tanner W. New potential cell wall glucanases of Saccharomyces cerevisiae and their involvement in mating. J Bacteriol 1998;180(19):5030-7.
Bensen ES, Filler SG, Berman J. A forkhead transcription factor is important for true hyphal as well as yeast morphogenesis in Candida albicans. Eukaryot Cell 2002;1(5):787-98. https://doi.org/10.1128/EC.1.5.787-798.2002.
Henar VM, Duran A, Roncero C. Chitin synthases in yeast and fungi. Experientia Suppl 1999;87:55-69.
Dunkler A, Walther A, Specht CA, Wendland J. Candida albicans CHT3 encodes the functional homolog of the Cts1 chitinase of Saccharomyces cerevisiae. Fungal Genet Biol 2005;42(11):935-947. https://doi.org/10.1016/j.fgb.2005.08.001.
Hazen BW, Hazen KC. Isolation of hydrophobic and hydrophilic variants of Candida albicans. Fems Microbiol Lett 1989;48(2):167-71.
https://doi.org/10.1111/j.1574-6968.1989.tb03293.x.
Singleton DR, Fidel PJ, Wozniak KL, Hazen KC. Contribution of cell surface hydrophobicity protein 1 (Csh1p) to virulence of hydrophobic Candida albicans serotype a cells. Fems Microbiol Lett 2005;244(2):373-7. https://doi.org/10.1016/j.femsle.2005.02.010.
Contreras-Shannon V, McAlister-Henn L. Influence of compartmental localization on the function of yeast NADP+ - specific isocitrate dehydrogenases. Arch Biochem Biophys 2004;423(2):235-46. https://doi.org/10.1016/j.abb.2003.12.038.
Zhao X, Oh SH, Yeater KM, Hoyer LL. Analysis of the Candida albicans Als2p and Als4p adhesins suggests the potential for compensatory function within the Als family. Microbiology 2005;151(Pt 5):1619-30. https://doi.org/10.1099/mic.0.27763-0.
Green CB, Cheng G, Chandra J, Mukherjee P, Ghannoum MA, Hoyer LL. RT-PCR detection of Candida albicans ALS gene expression in the reconstituted human epithelium (RHE) model of oral candidiasis and in model biofilms. Microbiology 2004;150(Pt 2):267-75. https://doi.org/10.1099/mic.0.26699-0.
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2017 Bosnian Journal of Basic Medical Sciences

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2016-12-09
Published 2017-05-20





